This is an old revision of the document!
Table of Contents
Gene prediction curation with IGV
Joran Martijn (February 2023)
After applying one or more gene prediction algorithms on your genome, you would do well to manually curate the predicted gene models manually. Even the most advanced algorithms to date can still make mistakes that are otherwise obvious to the human eye.
The curation process described here aims to compile all the necessary information in a single screen in the graphical user interface of the Integrated Genomics Viewer (IGV), which allows you to edit genes one by one if deemed necessary.
Example
An example view:
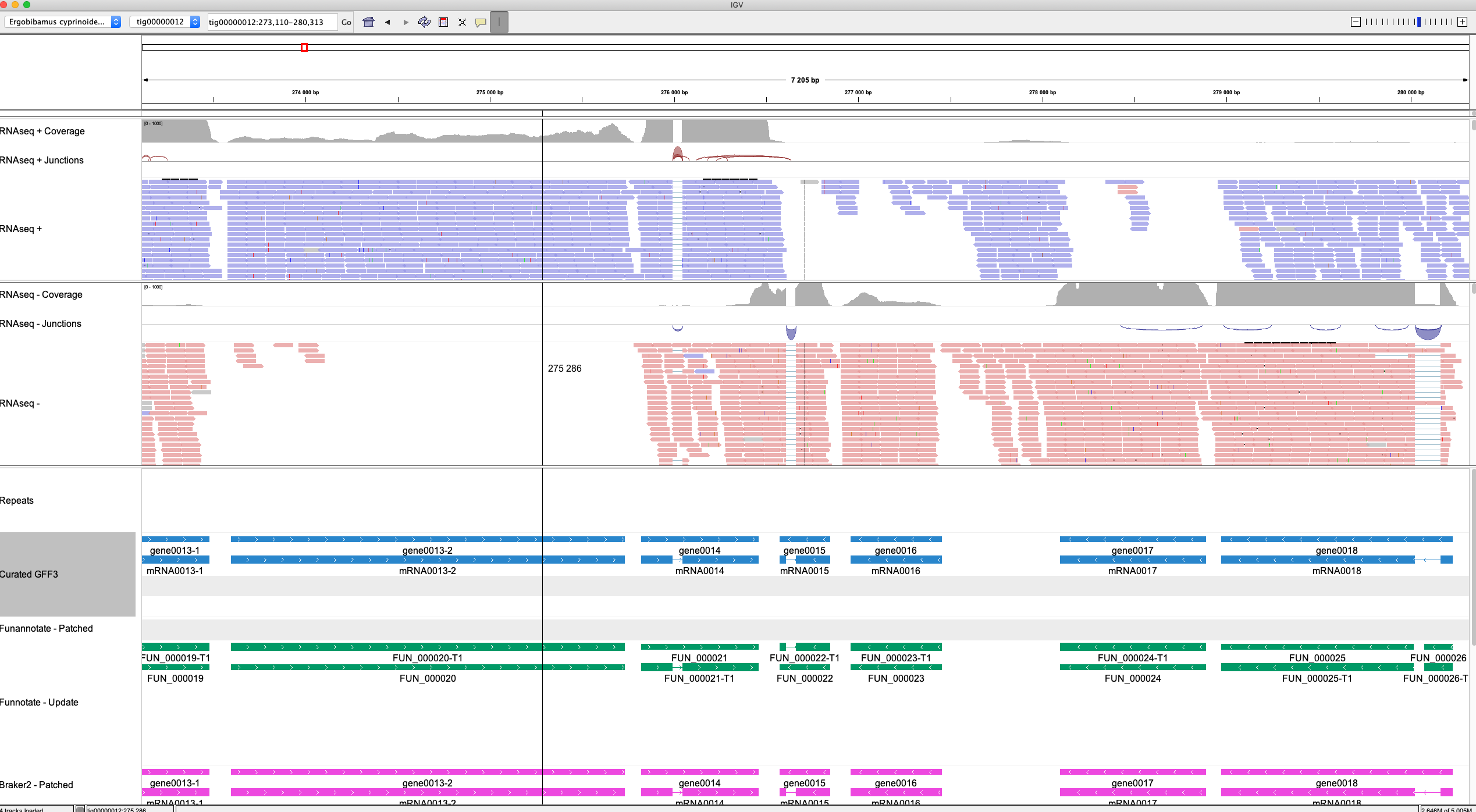
This example integrates the following source of information (top-to-bottom):
- Location in the genome
- RNAseq coverage levels
- RNAseq intron junctions
- The RNAseq reads mapped to the Ergobibamus genome sequence
- The Ergobibamus genome sequence (visible when zooming in)
- Repeats predicted by RepeatMasker
- The gene models predicted by Funannotate, Braker2 and various GeneMark algorithms (not visible in the screenshot)
Strand-specific RNAseq reads
Note that here two distinct RNAseq tracks are loaded. One for all the reads stemming from transcripts transcribed from the positive strand of the genome, and one for all those transcribed from the negative strand of the genome. This kind of strand-specificity information is only available when a RNAseq library protocol was used to retains this information, for example the dUTP method. I believe nowadays this is the default so it should be available to you.
If you used HISAT2 to map your RNAseq reads to your genome, all “positive” reads will have the tag XS:A:+, while all “negative” reads will have the XS:A:- tag. One can separate the two types of reads from the HISAT2-generated BAM file using the following command:
samtools view -b --tag XS:+ <rnaseq.bam> > <rnaseq.pos.bam> samtools index <rnaseq.pos.bam> <rnaseq.pos.bam.bai> samtools view -b --tag XS:- <rnaseq.bam> > <rnaseq.neg.bam> samtools index <rnaseq.neg.bam> <rnaseq.neg.bam.bai>
Loading into IGV
To load everything into a fresh IGV window, one can load each track in the old-fashioned way.
First load your genome with Genomes→Load Genome from File…, and then load each individual track with File→Load from File…. This is fine doing once, but if your IGV crashes for whatever reason, or becomes a tangled web of tracks or you accidentally removed your Sequence track (these things happen more frequently than you want) you will need to restart IGV and load everything from scratch, collapse/expand and color tracks as you desire, etc, etc again. This is a huge pain in the ass!
The second way would be to load everything manually as before, but then use File→Save Session… and reload sessions after crashing and restarting IGV.
The third, and my preferred way, is to load everything at once using a JSON file. See an example file below:
{
"id": "ergo",
"name": "Ergobibamus cyprinoides CL",
"fastaURL": "ergo_cyp_genome.fasta",
"indexURL": "ergo_cyp_genome.fasta.fai",
"chromosomeOrder": [
"tig00000012",
"tig00000492",
"tig00000016",
"tig00000015",
"tig00000496",
"tig00000019",
"tig00000498",
"tig00000497",
"tig00000079",
"tig00000031",
"tig00000018",
"tig00000013",
"tig00000513",
"tig00000510",
"tig00000506",
"tig00000491",
"tig00000445",
"tig00000507",
"tig00000505",
"tig00000509",
"tig00000514",
"tig00000511",
"tig00000092",
"tig00000512",
"tig00000494",
"tig00000493",
"tig00000495",
"tig00000508"
],
"tracks": [
{
"name": "Repeats",
"type": "annotation",
"format": "gff3",
"url": "repeats.gff3"
},
{
"name": "Curated GFF3",
"type": "annotation",
"format": "gff3",
"url": "01_curated_purged.gff3",
"color": "#2986cc",
"displayMode": "EXPANDED"
},
{
"name": "Funannotate - Patched",
"type": "annotation",
"format": "gff3",
"url": "funannotate_predict_patched.gff3",
"color": "#ff8200"
},
{
"name": "Funnotate - Update",
"type": "annotation",
"format": "gff3",
"url": "funnotate.gff3",
"color": "#009966",
"displayMode": "EXPANDED"
},
{
"name": "Braker2 - Patched",
"type": "annotation",
"format": "gff3",
"url": "curated_purged.contigs_012_016_492.gff3",
"color": "#ed48df"
},
{
"name": "Braker2 - Simplified",
"type": "annotation",
"format": "gff3",
"url": "EVM.raw.simple.gff3",
"color": "#f1c232",
"displayMode": "COLLAPSED"
},
{
"name": "GeneMark-ES",
"type": "annotation",
"format": "gff3",
"url": "genemark_ES.gtf",
"color": "#000000",
"displayMode": "COLLAPSED"
},
{
"name": "GeneMark-ET",
"type": "annotation",
"format": "gff3",
"url": "genemark_ET.gtf",
"color": "#bcbcbc",
"displayMode": "COLLAPSED"
},
{
"name": "RNAseq +",
"type": "alignment",
"format": "bam",
"url": "rnaseq_vs_masked_ergo_cyp_genome.sort.pos_transcripts.bam",
"indexURL": "rnaseq_vs_masked_ergo_cyp_genome.sort.pos_transcripts.bam.bai"
},
{
"name": "RNAseq -",
"type": "alignment",
"format": "bam",
"url": "rnaseq_vs_masked_ergo_cyp_genome.sort.neg_transcripts.bam",
"indexURL": "rnaseq_vs_masked_ergo_cyp_genome.sort.neg_transcripts.bam.bai"
}
]
}
This has some advantages:
- Load everything in one go
- Set up a desired display order of contigs / chromosomes
- Set up “clean” names for each track
- Set up the colors for each track
- Easy to share as all settings are in a plain text file
- You could automatically generate such a file in a pipeline if you want
Unfortunately I haven't been able to get it to collapse / expand tracks. In theory it should work with “displayMode”: “COLLAPSED” or “displayMode”: “EXPANDED” but it doesn't seem to work. In addition, you would still need to collapse the RNAseq intron junction track and the coloring of the RNAseq reads manually.
"name": Desired display name for your track "type": Type of track. Could be "alignment", "annotation" and perhaps other types "format" : "bam" or "gff3" "url" : filename of the file you wish to load here "fastaURL" : filename of the genome FASTA file "indexURL" : name of the indexed BAM file (BAI) or FASTA file (FAI) "color" : hexadecimal code of color you wish to use for the track
Adjust graphical settings
To color the RNAseq reads according to strandness, right-click on the track, then Color alignments by→first-of-pair strand
It may be useful to set a fixed y-scale of the coverage track. To do so, right-click on the coverage track, then Set Data Range, and enter your desired maximum value. I like 1000
Navigating IGV
I highly recommend using hotkeys to navigate IGV as much as possible. Once you have selected a track on the left (by clicking on it), you can:
+ Zoom in - Zoom out Ctrl+F Move to the next gene feature Ctrl+B Move to the previous gene feature Arrows Move a bit to the left or a bit to the right
For some frustratingly unknow reason, IGV only displays the three forward frames, OR the three reverse frames. It is, as far as I know, not possible to display all six frames simultaneously. To swap between reverse and forward frames, click on small arrow of the Sequence track. To see the sequence track, you need to be sufficiently zoomed in.
Curating gene models
IGV is excellent for viewing your data, but unfortunately not so much for editing your files in place. My solution has been to have the GFF3 file with your gene predictions open in a separate window, and adjusting the coordinates / adding or removing introns and exons directly in the text file. To make your changes visible in IGV, you need to remove the track and reload it. To make editing GFF3 files easier, I strongly recommend organizing your GFF3 files such that there is an empty line between each gene.
For example:
tig00000016 EVM gene 1991975 1995790 . - . ID=ctg016.gene0736;Name=gene0736 tig00000016 EVM mRNA 1991975 1995790 . - . ID=ctg016.mRNA0736;Parent=ctg016.gene0736;Name=mRNA0736 tig00000016 EVM CDS 1991975 1995790 . - 0 ID=ctg016.mRNA0736.CDS01;Parent=ctg016.mRNA0736 tig00000016 EVM exon 1991975 1995790 . - . ID=ctg016.mRNA0736.exon01;Parent=ctg016.mRNA0736 tig00000016 Manual gene 1995957 1996271 . - . ID=ctg016.gene0737-1;Name=gene0737-1 tig00000016 Manual mRNA 1995957 1996271 . - . ID=ctg016.mRNA0737-1;Parent=ctg016.gene0737-1;Name=mRNA0737-1 tig00000016 Manual CDS 1995957 1996271 . - 0 ID=ctg016.mRNA0737-1.CDS;Parent=ctg016.mRNA0737-1 tig00000016 Manual exon 1995957 1996271 . - . ID=ctg016.mRNA0737-1.exon01;Parent=ctg016.mRNA0737-1 tig00000016 Manual gene 1996590 1997048 . - . ID=ctg016.gene0737-3;Name=gene0737-3 tig00000016 Manual mRNA 1996590 1997048 . - . ID=ctg016.mRNA0737-3;Parent=ctg016.gene0737-3;Name=mRNA0737-3 tig00000016 Manual CDS 1996590 1997048 . - 0 ID=ctg016.mRNA0737-3.CDS;Parent=ctg016.mRNA0737-3 tig00000016 Manual exon 1996590 1997048 . - . ID=ctg016.mRNA0737-3.exon01;Parent=ctg016.mRNA0737-3
Curation example
Below we see an example of a potential gene that was missed by the gene prediction algorithm on the reverse strand. By checking the sequence strand and carefully moving from potential exon to exon, we can see that there is a potential gene here.

Using the ruler line of IGV (toolbar on the top, the last button on the right), we can determine the right coordinates, and we add this gene in our curated GFF3 file:
tig00000015 funannotate gene 617629 617991 . - . ID=ctg015.gene001388-2f;Name=gene001388-2f; tig00000015 funannotate mRNA 617629 617991 . - . ID=ctg015.mRNA001388-2f;Parent=ctg015.gene001388-2f;Name=mRNA001388-2f tig00000015 funannotate CDS 617629 617785 . - 1 ID=ctg015.mRNA001388-2f.cds;Parent=ctg015.mRNA001388-2f tig00000015 funannotate exon 617629 617785 . - . ID=ctg015.mRNA001388-2f.exon03;Parent=ctg015.mRNA001388-2f tig00000015 funannotate CDS 617835 617886 . - 2 ID=ctg015.mRNA001388-2f.cds;Parent=ctg015.mRNA001388-2f tig00000015 funannotate exon 617835 617886 . - . ID=ctg015.mRNA001388-2f.exon02;Parent=ctg015.mRNA001388-2f tig00000015 funannotate CDS 617937 617991 . - 0 ID=ctg015.mRNA001388-2f.cds;Parent=ctg015.mRNA001388-2f tig00000015 funannotate exon 617937 617991 . - . ID=ctg015.mRNA001388-2f.exon01;Parent=ctg015.mRNA001388-2f
Note that I didn't explicitly defined intron features here, only gene, mRNA, CDS and exon features. The introns are implied as the coordinate space between exons. You can also explicitly define them if you want. IGV will work with either formatting choice.
Reload the Curated GFF3 track with the updated GFF3 file:

Also zoom in until you see the protein sequence to make sure that you haven't accidentally introduced any frameshifts (usually visible by premature STOP codons)
GFF3 Format
To edit GFF3 files, you must have a good understanding of the format. GFF3 is an extension of the older GTF format and an attempt to standardize the gene annotation format. The official rules and guidelines can be found on their GitHub page: GFF3 Format. This is quite a long and extensive page. For our purposes, we'll mainly need to know the following things:
- GFF3 files are tab-delimited, and consist of 9 columns
- Each line defines 1 feature. For example, gene, mRNA, exon, CDS, intron, etc
The 9 columns are as follows:
| Column | Description |
|---|---|
| SeqID | Name of contig, chromosome, etc |
| Source | Source of the feature. E.g. genemark, braker, etc |
| Type | gene, mRNA, exon, intron, CDS etc |
| Start | Start coordinate |
| End | End coordinate |
| Score | Some kind of score. . if not defined |
| Strand | + or - |
| Phase | 0, 1, or 2. Only relevant for CDS features |
| Attributes | tag=value; tag=value; See below |
Phase is not the same as the reading frame. In short,
0 means that the first available codon starts on the 1st base of the feature
1 means that the first available codon starts on the 2nd base of the feature
2 means that the first available codon starts on the 3rd base of the feature
Thus, the CDS corresponding to the first exon always has phase 0. CDSs of subsequent exons can have phases 0, 1 or 2.
For example, if the second exon of a gene has sequence CGTTACAGAGC…, and the CDS has phase 1, the first codon of this sequence starts at the G, i.e. C-GTT-ACA-GAG-C…. However, if it had phase 2, it would be CG-TTA-CAG-AGC